In this project, you will implement dynamic programming algorithms for computing the minimal cost of aligning gene sequences and for extracting optimal alignments.
You are using dynamic programming to align multiple gene sequences (taxa), two at a time. In light of the SARS outbreak a few years ago, we have chosen to use the SARS virus as one of our DNA sequences. SARS is a coronavirus, and we have also provided the genomes for several other coronaviruses. It is your job in this project to align all pairs in order to assess the pair-wise similarity. To prepare to succeed on this project, make sure you understand the sequence alignment and solution extraction algorithms as presented in class and in the book.
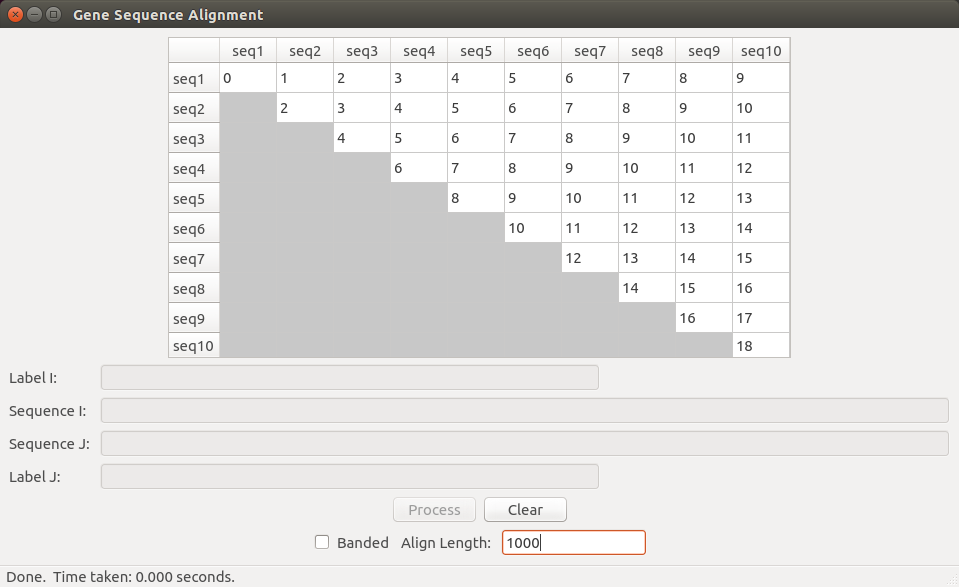
You are provided with some scaffolding code to help you get started on the project. We provide a PyQt GUI containing a 10x10 matrix, with a row and a column for each of 10 organisms. The organism at row i is the same organism for column i. ***Note that this matrix is not the dynamic programming table***; it is simply a place to store and display the final result from the alignment of the gene sequences for each pair of organisms. Thus, cell (i, j) in this table will contain the minimum cost alignment score of the genome for organism i with the genome for organism j.
When you press the “Process” button on the GUI, the matrix fills with numbers, one number for each pair, as shown in the figure below. You will generate these numbers by aligning the first n characters (bases) in each sequence pair (the default will be n = 1000 but you can change this). Note that your alignment may be slightly longer than this due to inserts. Your job will be to fill in the proper numbers based on a sequence alignment function that employs dynamic programming. You will fill the matrix with the pair-wise scores. You do not need to fill in the lower triangle of the matrix (since it is symmetric), but you should fill in the diagonal. When the “Process” button is clicked, the GUI calls the GeneSequencing.align_all() method which you will implement.
Each gene sequence consists of a string of letters and is stored in the given database file. The scaffolding code loads the data from the database. For example, the record for the “sars9” genome contains the following sequence (prefix shown here):
atattaggtttttacctacccaggaaaagccaaccaacctcgatctcttgtagatctgttctctaaacgaactttaaaatctgtgtagctgtcgctcggctgcatgcctagtgcaccta...
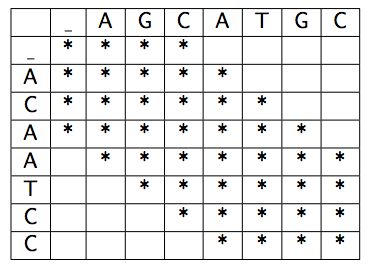
Each algorithm should use dynamic programming: in particular, use the Needleman/Wunsch cost function, which we discussed in class and that is shown below, to compute the distance between two sequences. Find the best alignment by minimizing the total cost.
The requirement is that your unrestricted algorithm run in at most O(nm) time and space, as discussed in class and the text, where n and m are the lengths of the two sequences. Your banded algorithm should should run in at most O(kn) time and O(kn) space, where k is the bandwith and n is the length of the shorter sequence .
You must fill the 10x10 results matrix for 1000 base pairs with your unrestricted algorithm in less than 120 seconds using O(nm) time and space. Remember: Since the matrix is symmetric, you should only fill the upper part of the matrix (above the diagonal).
Your banded algorithm should fill in the same matrix for 3000 base pairs in less than 10 seconds using O(kn) time and space, where k is your bandwidth constant and n is the length of the shorter sequence.
Write a report describing your work containing the following elements:
Here are a couple of suggestions for further exploration, for any who might be interested. They are listed without prejudice against things that are or are not on this list.
The following explains the biological setting of this project, including some background on SARS and Coronaviruses in general from the Department of Microbiology and Immunology, University of Leicester.
Coronaviruses: Coronaviruses were first isolated from chickens in 1937. After the discovery of Rhinoviruses in the 1950’s, approximately 50% of colds still could not be ascribed to known agents. In 1965, Tyrell and Bynoe used cultures of human ciliated embryonal trachea to propagate the first human coronavirus (HCoV) in vitro. There are now approximately 15 species in this family, which infect not only man but cattle, pigs, rodents, cats, dogs, and birds (some are serious veterinary pathogens, especially in chickens).
Coronavirus particles are irregularly-shaped, ~60-220nm in diameter, with an outer envelope bearing distinctive, ‘club-shaped’ peplomers (~20nm long x 10nm at wide distal end). This ‘crown-like’ appearance (Latin, corona) gives the family its name. The center of the particle appears amorphous in negatively shaped stained EM preps, the nucleocapsid being in a loosely wound rather disordered state. Most human coronaviruses do not grow in cultured cells, therefore relatively little is known about them, but two strains grow in some cell lines and have been used as a model. Replication is slow compared to other envelope viruses, e.g. influenza.
Coronavirus infection is very common and occurs worldwide. The incidence of infection is strongly seasonal, with the greatest incidence in children in winter. Adult infections are less common. The number of coronavirus serotypes and the extent of antigenic variation is unknown. Re-infections occur throughout life, implying multiple serotypes (at least four are known) and/or antigenic variation, hence the prospects for immunization appear bleak.
SARS: SARS is a type of viral pneumonia, with symptoms including fever, a dry cough, dyspnea (shortness of breath), headache, and hypoxaemia (low blood oxygen concentration). Typical laboratory findings include lymphopaenia (reduced lymphocyte numbers) and mildly elevated aminotransferase levels (indicating liver damage). Death may result from progressive respiratory failure due to alveolar damage. The typical clinical course of SARS involves an improvement in symptoms during the first week of infection, followed by a worsening during the second week. Studies indicate that this worsening may be related to a patient’s immune responses rather than uncontrolled viral replication.
The outbreak is believed to have originated in February 2003 in the Guangdong province of China, where 300 people became ill, and at least five died. After initial reports that a paramyxovirus was responsible, the true cause appears to be a novel coronavirus with some unusual properties. For one thing, SARS virus can be grown in Vero cells (a fibroblast cell line isolated in 1962 from a primate)---a novel property for HCoVs, most of which cannot be cultivated. In these cells, virus infection results in a cytopathic effect, and budding of coronavirus-like particles from the endoplasmic reticulum within infected cells.
Amplification of short regions of the polymerase gene, (the most strongly conserved part of the coronavirus genome) by reverse transcriptase polymerase chain reaction (RT-PCR) and nucleotide sequencing revealed that the SARS virus is a novel coronavirus which has not previously been present in human populations. This conclusion is confirmed by serological (antigenic) investigations. We now know the complete ~29,700 nucleotide sequence of many isolates of the SARS virus. The sequence appears to be typical of coronaviruses, with no obviously unusual features, although there are some differences in the make up of the non-structural proteins which are unusual.
There is currently no general agreement that antiviral drugs have been shown to be consistently successful in treating SARS or any coronavirus infection, nor any vaccine against SARS. However, new drugs targeted specifically against this virus are under development.
Coronaviruses with 99% sequence similarity to the surface spike protein of human SARS isolates have been isolated in Guangdong, China, from apparently healthy masked palm civets (Paguma larvata), a cat-like mammal closely related to the mongoose. The unlucky palm civet is regarded as a delicacy in Guangdong and it is believed that humans became infected as they raised and slaughtered the animals rather than by consumption of infected meat.
Might SARS coronavirus recombine with other human coronaviruses to produce an even more deadly virus? Fortunately, the coronaviruses of which we are aware indicate that recombination has not occurred between viruses of different groups, only within a group, so recombination does not seem likely given the distance between the SARS virus and HCoV.
SARS, The Disease and the Virus (from The National Center for Biotechnology Information): In late 2002, an outbreak of severe, atypical pneumonia was reported in Guangdong Province of China. The disease had an extremely high mortality rate (currently up to 15-19%), and quickly expanded to over 25 countries. The World Health Organization coined it "severe acute respiratory syndrome", or SARS. In April 2003, a previously unknown coronavirus was isolated from patients and subsequently proven to be the causative agent according to Koch's postulates in experiments on monkeys. The virus has been named SARS coronavirus (SARS-CoV).
The first complete sequence of SARS coronavirus was obtained in BCCA Genome Sciences Centre, Canada, about two weeks after the virus was detected in SARS patients. It was immediately submitted to GenBank prior to publication as a raw nucleotide sequence. GenBank released the sequence to the public the same day under accession number AY274119; the NCBI Viral Genomes Group annotated the sequence also the same day and released it in the form of the Genomes RefSeq record NC_004718 at 2 am next day. As of the beginning of May of 2003, all the SARS-CoV RNA transcripts have been detected and sequenced in at least two laboratories; further experiments are underway.
The availability of the sequence data and functional dissection of the SARS-CoV genome is the first step towards developing diagnostic tests, antiviral agents, and vaccines.